Protein folding, protein dynamics, protein structure/function
relationships, protein-protein interactions and polymerization/aggregation
mechanisms are projects that we have studied in this laboratory.
Protein Folding
The long term goal of the protein folding studies was to understand the
nature of the unfolded and intermediate structures on the unfolding and
refolding pathways, including the role of proteins that assist folding
(called chaperonins). The work uses site-directed mutagenesis and
techniques such as 19F and proton NMR, circular dichroism,
fluorescence measurements and x-ray crystallography. Current studies
include work with the apoE family of proteins, with the role of proline in
protein folding and with intrinsically disordered proteins such as amyloid
beta and the bacterial protein CsgA.
Many of the studies involved incorporating fluorine labeled amino acids
into the protein and then examining the NMR spectrum. We use stopped flow
methods in conjunction with a fluorine cryoprobe for these measurements.
From data collected in current and past projects we have a large database
of fluorine chemical shifts in proteins. These shifts are sensitive to low
concentrations of denaturant, to temperature and to pH. A website for
fluorine chemical shifts has been established at
https://biochem.wustl.edu/~bmbnmr/Fluorine.html.
Past Projects
The goal of this work is to understand the GroEL-mediated
folding mechanism. GroEL
is a member of the Hsp60 class of chaperones,
which are tetradecamers of identical 57.2 kDa monomers. The chaperonin
binds unfolded proteins and generally increases the final yield of native
protein without increasing the rate of folding. The formation of stable
complexes with GroEL is most often discussed, and while, for example,
murine dihydrofolate reductase (MuDHFR) does form a stable complex, the
complex formed with the structurally homologous E. coli DHFR
(EcDHFR) is transient (at 22 degrees C). For both DHFRs, the concentration
of bound
protein increases as the temperature is increased. Using a variety of
biophysical approaches, we are examining the
kinetic mechanism of folding as well as the thermodynamic parameters for
the binding of these DHFRs with GroEL. Our most recent work examines
the role of ligands such as K+, Mg2+, ATP
and GroES on the folding mechanism.
To read recent abstracts of this work, click Here.
|
|
PapD is a protein required for the formation of pili in pathogenic
bacteria. It, however, does not get incorporated into the pilus and is
believed to function as a chaperone for the folding of other proteins
which make up the pilus structure. This protein is rich in proline and has
two distinct domains. The folding is sufficiently slow that we can apply
NMR techniques to examine the folding process much like have been used
with dihydrofolate reductase (see above). Currently we are examining the
role of domain-domain interaction on the folding process.
To read recent abstracts of this work, click Here
|
The goal of this work is to understand the molecular mechanism of actin
polymerization and filament function. We are investigating the role of
specific amino acid residues in the mechanism by comparing the kinetics of
self- polymerization of mutant actin proteins with that of wild type, in
the presence and absence of various actin binding proteins. Because yeast
genes are more readily manipulated than those of higher eucaryotes, we are
primarily studying yeast actin including several actin mutations that have
been phenotypically characterized in nematodes which we have put into
yeast actin.
To read recent abstracts of this work, click
Here.
|
|
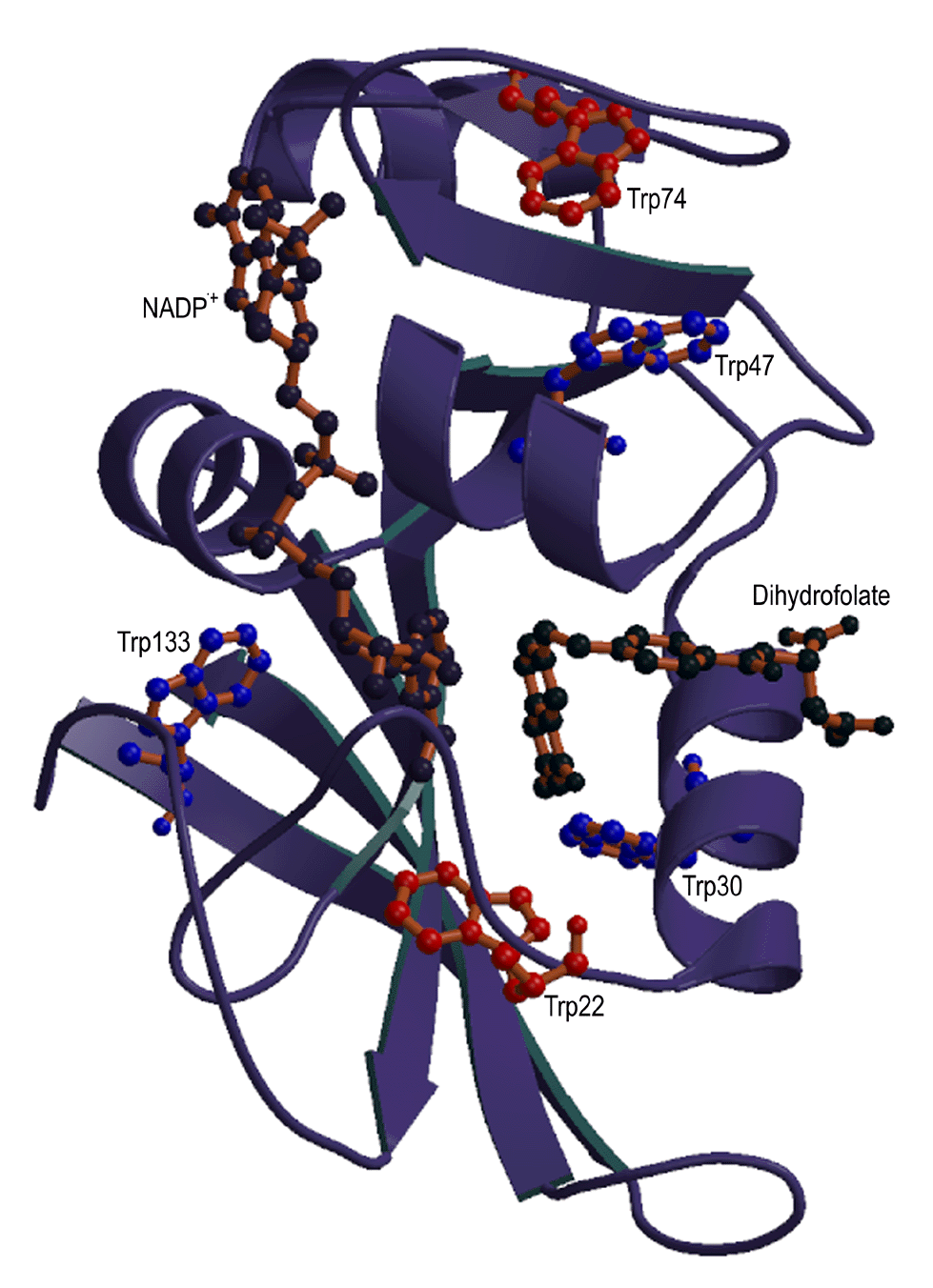
In this work, we are studying sidechain environment and behavior
during the refolding of E. coli dihydrofolate reductase (DHFR) in real time by stopped-flow NMR techniques.
E. coli dihydrofolate reductase has five tryptophans which have
been replaced with 6-19F-tryptophan. The resonances
assigned to each tryptophan are resolved in both unfolded and native
DHFR, allowing us to monitor the environment of individual tryptophans
during unfolding and refolding in real time using stopped-flow
19F NMR techniques. This allows, for the first time,
measurements of stabilization of specific regions of a protein during
the refolding process either in the presence or absence of ligands. The
work is being extended using 19F-labeled phenylalanine as markers
for specific region or domain stabilization during refolding. The
refolding and unfolding kinetics have also been examined by
stopped-flow circular dichroism and stopped-flow fluorescence techniques and compared
to the sidechain environment observed by stopped-flow 19F NMR.
To read recent abstracts of this
work click Here.
|
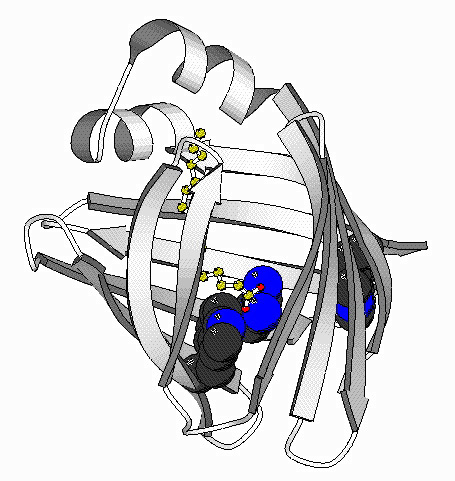
Folding Studies of Intestinal Fatty Acid Binding Protein
|
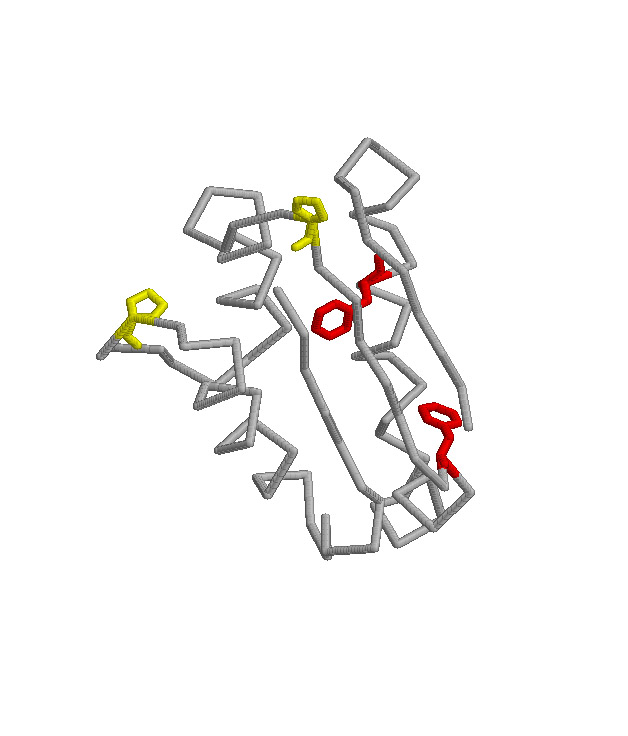
The goal of this project is to determine the mechanism of folding of the
intestinal fatty acid binding protein (IFABP). This
protein is one of a family of proteins that bind fatty acids, bile salts
and retinoids. This family is primarily beta-sheet with
a large central cavity into which the ligand binds. Cysteine-
and proline-free, IFABP is a model protein for studying the
mechanism of folding.The lack ofproline allows us to explore the role of
proline in the folding process. We have developed the technique of turn
scanning (mutagenesis in turns) to examine the role of turns in the folding
process. By NMR, we are currently examining structrual changes that occur
throughout the molecule as a consequence of single site mutations. We are
also using this protein to explore the method of fluorescence correlation
spectroscopy (FCS) in combination with fluorescence resonance energy
transfer (FRET) as it applies to protein structural changes in both the
native and unfolded states.
To read recent abstracts of this work, click
Here.
|
|
Work with the murine (mouse) adenosine deaminase continues our attempt to
understand the folding of larger and larger proteins. Adenosine deaminase
has a molecular weight of 40 kD and is present in virtually all mammalian
cells. It is a key enzyme in purine metabolism. Lack of enzymatic
activity, leading to severe T, NK and B lymphocytopenia, is associated
with about 20-30 percent of children with severe combined immunodeficiency
(SCID). In addition it contains a tightly bound Zn atom that is essential
for enzymatic activity. There are numerous reports in the literature of
mutations that lead to the loss of enzymatic activity associated with
SCID. Some, understandably, are in the active site of the enzyme while
others, surprisingly, are distant from the active site. We will study why
distant mutations lead to partial or total loss of enzymatic activity. We
suggest that a possible explanation for the loss of activity in these
distant mutations arises either from large conformational changes or from
a loss of ability of the protein to fold properly. The studies involve
expression of wild-type and mutant murine adenosine deaminases (the murine
enzyme is highly homologous to the human enzyme) followed by
characterization of the properties using fluorescence, circular dichroism
and NMR techniques as well as enzymatic activity. In particular we are
interested in the characterization of folding properties and the role of
Zn in folding.
To read recent abstracts of this work, click Here
|
Dr. Carl Frieden
Department of Biochemistry and Molecular Biophysics, Box 8231
Washington University School of Medicine
660 South Euclid
St. Louis, MO 63110 (USA)
office: 314-362-3344
lab: 314-362-3342
or -3359
FAX: 314-362-7183
send mail to frieden@biochem.wustl.edu
URL: https://biochem.wustl.edu/faculty/frieden
last updated: 4/3/18
|
|